






This paper summarizes and draws up the decision tree for the in vitro consistency evaluation of oral solid generic drugs in combination with the relevant technical requirements such as “Requirements for Application Materials for Consistency Evaluation of the Quality and Efficacy of Oral Solid Preparations of Chemical Generic Drugs (Trial)”, and discusses the in vitro consistency evaluation under different circumstances. To evaluate the differences and common problems of inspection projects, focus on analyzing the technical requirements and concerns of unconventional research projects, and put forward corresponding treatment suggestions, aiming to provide more references for subsequent studies on consistency evaluation of oral solid generic drugs.
Significant progress has been made in the consistency evaluation of oral solid preparations since its official launch in 2017. The overall research level of the declared varieties has been significantly improved, mainly reflected in the improvement of in vitro evaluation research, the gradual reduction of the issue of supplements, and the continuous increase in the rate of passing the consistency evaluation. As a supporting document of the newly promulgated “Administrative Measures for Drug Registration”[1], “Chemical Drug Registration Classification and Application Materials Requirements (Draft for Comment)”[2] once again clearly requires that the generic drugs of category 3, 4 and 5.2 should be consistent with the quality and efficacy of the reference preparation. Consistency evaluation is no longer a phased work, but a basic requirement for the listing of generic drugs, which runs through the marketing application, and the relevant requirements are more refined. This article combines the “Notice on Issuing the Application Materials Requirements for the Quality and Efficacy Consistency Evaluation of Chemical Drug Generic Drugs (Trial)” (2016 No. 120) [3], “Announcement on Matters Concerning the Quality and Efficacy Consistency Evaluation of Generic Drugs” (2017 No. 100) [4] and related requirements, a decision tree for in vitro consistency evaluation of oral solid generic drugs was drawn (Figure 1), and based on this, through representative cases, it focused on analyzing unconventional research projects. The technical requirements and concerns were raised, and corresponding suggestions were put forward.
At the same time, the focus and common problems of in vitro evaluation research under special circumstances such as applying for bioequivalent (BE) exempted varieties and varieties that could not be recommended for reference preparations were discussed. The purpose is to provide more reference for the subsequent application of oral solid generic drugs. In vitro evaluation is listed separately as an important part in the consistency evaluation, which shows its importance. Although there are no clear requirements for the newly registered generic drugs, it runs through the entire research process such as formulation process development, quality research and stability inspection. The in vitro evaluation is carried out on the basis of determining the rationality of the selection of reference preparations, including two main parts: quality consistency evaluation and dissolution curve similarity evaluation.
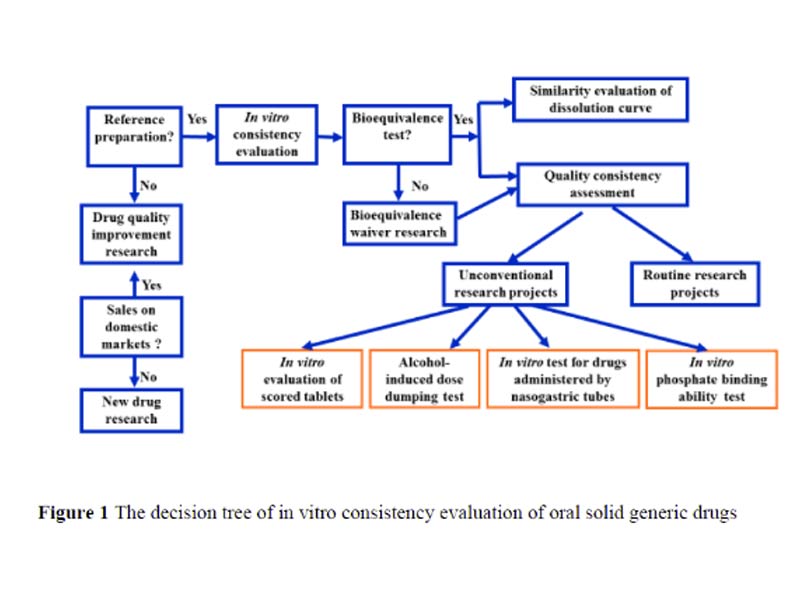
Quality Consistency Evaluation
1.1 Research On Routine Projects
Items included in domestic and foreign pharmacopoeia standards are usually routine and must be studied, and generally include key quality attributes such as related substances, content, dissolution and content uniformity. For chiral drugs, isomer impurities should also be studied, and polymorphic drugs should be studied for the type of crystal form or the purity of crystal form [5]. It is generally required to list the test data of multiple batches of self-made products and multiple batches of reference preparations, evaluate and analyze the quality consistency of self-made products and reference preparations, and provide detailed impurity spectrum analysis, and pay attention to the degradation trend of unknown impurities with large detected values in the subsequent stability investigation.
1.2 Research Based On Product Characteristics
The usage and dosage, mode of administration, in vivo drug release characteristics and action site of some reference preparations are special, such as the reference preparation has functional nicks, and the drug release in the body has the risk of ethanol-induced dose dumping, which can be passed through the nose. Gastric tube administration and intestinal topical administration are not easy to conduct in vivo BE studies, etc. These characteristics have an important impact on the quality controllability, safety and efficacy of the drug. For the substitutability in clinical use, the generic drugs also need to carry out corresponding design and quality research. The following section analyzes the technical requirements and concerns of these unconventional research projects based on domestic and foreign guidelines and specific cases.
1.2.1 Design And Consistency Study Of Functional Scoring Sheet
When studying the quality characteristics of the reference preparation, attention should also be paid to whether the tablet has nicks, and whether it is a functional nick should be judged according to the usage and dosage description on the instructions. For example, the 25 mg captopril tablet has a cross notch on one side, and the original research instructions clearly indicate the usage of half and 1/4 of the tablet; some sustained-release tablets, such as isosorbide mononitrate sustained-release tablets, have a groove on the side, which is described in the instruction manual. “The tablet can be broken along the groove, and half a tablet can be taken.” Similar to the above-mentioned nicks with the function of metering and dividing, they are all functional nicks. In order to fully ensure the replaceability in clinical use, the imitation tablet should be designed with the same functional notches, and the quality consistency and stability of the split part and the whole tablet should be studied. If the reference product does not have a functional score design, it is not recommended to design a functional score for the generic tablet [6]. The shape and size of the tablet, the design of the notch, and the way of dividing may all affect the quality of the divided part. Reference should be made to the “Guidelines for Design and Research on Functional Notch Design and Research on Oral Tablets of Chemical Generic Drugs” [7], and combined with the relevant information of the reference preparation to design a notch with accurate segmentation function. In the process of product development, scale-up research and verification, in vitro evaluation research of the segmented part should be carried out. The inspection items usually include weight difference, content uniformity, segmentation weight loss. For friability and dissolution, sustained-release preparations should also take the split part and the whole tablet at the upper and lower limits of the proposed whole tablet hardness range for similarity study of dissolution curves. If the research shows that the quality of the score sheet meets the requirements, these in vitro evaluation items may not be used as routine inspection items.

1.2.2 Consistency Study Of Ethanol-Induced Dose Dumping
Oral modified-release preparations are controlled by a polymer matrix or coating to control the release of the API. If taken together with alcoholic beverages, the controlled-release polymer may be dissolved or denatured in advance, resulting in excessive drug release and dose dumping. This in turn affects the safety and efficacy of drugs, especially drugs with a narrow therapeutic window and even causes death. Therefore, for generic drugs, the EMA guideline [8] requires that all oral modified-release preparations be subjected to a mid-dose dumping test in ethanol. The FDA Bioequivalence Division requires ethanol mid-dose dumping studies for all modified-release opioid formulations and other modified-release formulations that are prone to risk of dose dumping. FDA specific drug bioequivalence guidelines identify the types and specific methods that need to be tested for dose dumping, such as hydromorphone hydrochloride extended-release tablets [9], metoprolol succinate extended-release tablets [10] and venla hydrochloride Oral modified-release preparations such as Faxin sustained-release tablets [11]. For oral modified-release preparations, the ethanol mid-dose dumping test is actually a comparative study on the consistency of the dissolution/release profiles of the generic drug and the reference preparation under the conditions of different concentrations of ethanol in the dissolution medium (high rotation speed if necessary) to evaluate the performance of the generic drug in the possibility of sudden drug release in the in vivo alcohol environment and whether the release behavior is similar to that of the reference preparation. If the generic drug release is accelerated and the rate is not similar to the reference drug, the formulation should be re-formulated. In some specific cases, a BE test when the preparation is taken with alcohol may be required [12]. In fact, ethanol-induced dose dumping in vivo is affected by many factors, and the in vitro evaluation of the correlation with complex factors in vivo is a concern. EMA and FDA provide ethanol-induced dose dumping test methods, but the test conditions are not completely consistent. For example, FDA requires 40% ethanol concentration to be investigated, and EMA does not specify sampling time. Therefore, factors such as the properties of raw materials and excipients, formulation design, indications, assessment of the risk of dose dumping and the characteristics of the reference preparation should be comprehensively considered to select test conditions such as ethanol concentration, rotation speed and sampling time interval.
1.2.3 Consistency Study Of Nasogastric Tube Administration
Some original oral solid preparations, such as tegrilol tablets, rivaroxaban tablets, gefitinib tablets and lansoprazole delayed release orally disintegrating tablets, can be administered through nasogastric tube for patients who cannot swallow the whole tablet, that is, the tablets can be crushed and mixed with diluents such as water, glucose solution, jam and puree and then administered through nasogastric tube [13,14]. Compared with oral administration of the whole tablet, nasogastric tube administration may reduce the actual dosage, affect the state of drugs entering the stomach (such as sedimentation volume, particle size distribution, etc.), and then affect the dissolution or release of drugs, as well as the bioavailability and curative effect in vivo; At the same time, the tablets are ground into suspension, which will also increase the risk of mass instability. Therefore, the in vitro comparative evaluation of nasogastric tube administration of generic drugs and reference preparations is an important content of in vitro consistent evaluation. The investigation items usually include sedimentation test, particle size distribution, recovery rate and stability. When designing the test, you can refer to the methods recommended by FDA drug guidelines and conduct risk assessment in combination with the characteristics of the product. The selected test conditions should cover the worst conditions. For the release or absorption of drugs greatly affected by pH value, in vitro comparative tests should be carried out with media with different pH values. For example, the draft of individual drug guidelines for lansoprazole delayed release orally disintegrating tablets [15] requires tests such as disintegration time limit, sedimentation volume and recovery in media with different pH values (5.5, 7.0 and 8.5).

1.2.4 Consistency Study Of Phosphorus Binding Capacity In Vitro
Phosphate binders are commonly used drugs to reduce blood phosphorus concentration. There are many factors that affect the binding ability of such preparations to phosphate radicals in the gastrointestinal tract, including drug dissolution and molecular ion form, phosphate radical concentration, and pH of the gastrointestinal tract. Therefore, similar dissolution profiles do not mean the same phosphorus binding capacity. At the same time, considering that such preparations have local effects on the gastrointestinal tract, are not absorbed by oral administration, and are not easy to perform BE tests, in vitro phosphorus-binding capacity tests (ie, phosphorus-binding kinetics test and phosphorus-binding balance test) are usually recommended as a method of the equivalence evaluation. At present, there is no guiding principle for the investigation of phosphorus binding capacity at home and abroad. The FDA guideline recommends the method of phosphorus binding capacity test for such preparations, such as lanthanum carbonate chewable tablets [16], sevelamer carbonate tablets [17] and citrate Iron acid flakes [18] and so on. Reference [19] reviews the relevant requirements and summarizes the issues that need attention. The phosphorus binding ability test can be carried out with reference to the above data, and factors that may affect the binding of phosphorus binding agents to phosphate should be considered.
First, the formulation composition and process can affect the combination of the drug with the phosphate radical in the gastrointestinal tract through factors such as tablet hardness, disintegration time limit, and gastrointestinal pH value. Sufficient screening research should be carried out on the basis of the original product.
Secondly, a reasonable phosphorus-binding capacity test plan should be designed to establish a good correlation between in vivo and in vitro. For example, when selecting media with different pH values, the pH value of each segment of the gastrointestinal tract should be reflected, and the pH value of the drug action site should be paid attention to. During the test, the pH value should be kept stable to reduce the influence on the maximum equilibrium concentration of phosphorus binding and the kinetic curve; the volume of the medium used for investigation should be adjusted according to different specifications; the chewable tablet should be swallowed whole and simulated after chewing (crushing). The effect of taking on phosphorus binding capacity, etc. At the same time, each drug guide requires at least 12 preparation units each for the self-made product and the reference preparation for testing. For varieties that may have batch differences, such as sevelamer carbonate tablets, the number of preparations selected should be able to support the final data analysis. statistically significant. It is usually recommended to select a specific analytical method such as ion chromatography, and conduct a comprehensive method validation. Pay attention to whether the validation range of linearity and accuracy covers the actual concentration of phosphorus binding. Necessary methods should be performed in media with different pH values.
Evaluation Of Dissolution Profile Similarity
2.1 Screening Of Dissolution Conditions
The dissolution conditions and detection methods of dissolution curve can refer to the established dissolution conditions and methods. On this basis, combined with the characteristics of products, the dissolution conditions with good discrimination can be selected. In order to make the insoluble drugs reach the “slot leakage condition”, it is usually considered to add surfactants, and the amount of surfactants that need to be added may also increase with the increase of specification. For example, rivaroxaban tablets have three specifications of 10, 15 and 20 mg. The FDA dissolution method database recommends that the dissolution medium of 10 mg is pH 4.5 acetate buffer of 0.2% sodium dodecyl sulfate (SDS), and the dissolution medium of 15 and 20 mg is pH 4.5 acetate buffer of 0.4% SDS [20]. If 20 mg be test is used to exempt 10 and 15 mg be test, the similarity of dissolution curve in 0.4% SDS dissolution medium shall be studied for 15 and 20 mg be test; The recommended SDS concentrations of 10 and 20 mg specifications are different. When selecting the dissolution medium, the difference of dissolution amount caused by the different concentration ratio of main drug and surfactant should be avoided. It can be considered to select two tablets of 10 mg specification and one tablet of 20 mg specification in the dissolution medium of 0.4% SDS [21], or 10 mg specification in the dissolution medium of 0.2% SDS and 20 mg specification in the dissolution medium of 0.4% SDS for the Similarity Study of dissolution curve.

2.2 Frequently Asked Questions About Dissolution Profile Studies
The dissolution profiles of multiple batches of self-made products have large intra-batch differences, while the dissolution profiles of the reference preparations have good intra-batch uniformity, indicating that there is a problem with the quality of the self-made products, and the formulation process should be improved. If the reference preparation also has the problem of large differences in the dissolution curve within the batch, it should also consider whether the selection of the dissolution conditions is appropriate. It can be improved by increasing the rotation speed; if the volume of the dissolution medium is too small, the poorly soluble drug cannot be fully dissolved, and the extracted solution contains undissolved drug particles, and the drug is gradually dissolved from the particles during the sample processing process, because the processing time is not completely consistent. This leads to the difference of the dissolution amount within the batch. This situation can be further studied by increasing the volume of the dissolution medium, and if necessary, the addition of surfactants can be considered for optimization. If there is a change in the approved product that may affect the dissolution behavior of the product, such as the crystal form or particle size of the API, the type, dosage or supplier of binders and disintegrants in the formulation, mixing, tableting and coating, in order to examine the consistency of product quality before and after the change, in addition to the inspection items of the quality standard, the similarity test of the dissolution curve of the changed product and the BE batch of self-made products in various media should be carried out. If the BE batch of self-made products is no longer within the validity period, it can also be compared with the dissolution curve data of the original BE batch of self-made products, provided that the dissolution conditions and methods have not been changed. significance. At the same time, the statistics, collation and presentation of the dissolution curve similarity research data are also very important, which can be collated and submitted by referring to the literature [22].
In Vitro Studies Of BE Exempted Varieties
BE exemptions are generally divided into two categories: one is be exemption based on biopharmaceutical classification system (BCS), and the other is be exemption between multiple specifications. In both cases, the similarity of dissolution curve in media with multiple pH values should be studied, but the concerns are different. The be exemption based on BCS classification should prove that the dissolution curve of multiple batches of self-made products and reference preparations in multiple media is similar, while the be exemption between multiple specifications needs to prove that the proposed exemption specification is similar to the dissolution curve of be specification in multiple media. Be exemption between multiple specifications shall meet the requirement that the prescription proportion of the specification to be exempted is similar to that of be specification.
For the judgment of “high active substances” and “the prescription proportion of non-equal changes in the proportion of active ingredients and inactive ingredients is similar”, the domestic guiding principle [12] does not give clear standards. Referring to the relevant guidelines of EMA and FDA [21,23], it can be understood that the content of highly active substances is less than 5% of the content of tablet core / capsule; For multi specification prescriptions with unequal proportion of active ingredients and inactive ingredients, if the change range of the proportion of inactive ingredients relative to the single dose prescription is within the allowable range of class II change in the dosage change of excipients (± 10%), it also belongs to the similar proportion of prescriptions. The change range of the inactive ingredients of the proposed exemption specification is calculated based on the prescription of be specification. First calculate the proportion of the inactive ingredients of the proposed exemption specification and be specification relative to the single dose prescription, and then calculate the difference between the two specifications. When there is more than one type of inactive change in the prescription, the change range of inactive ingredients shall be the sum of the absolute values of the changes of each inactive ingredient. Take the prescription of multi specification ordinary tablets as an example to illustrate the application of the standard of “similar prescription proportion”. The prescription is shown in Table 1.
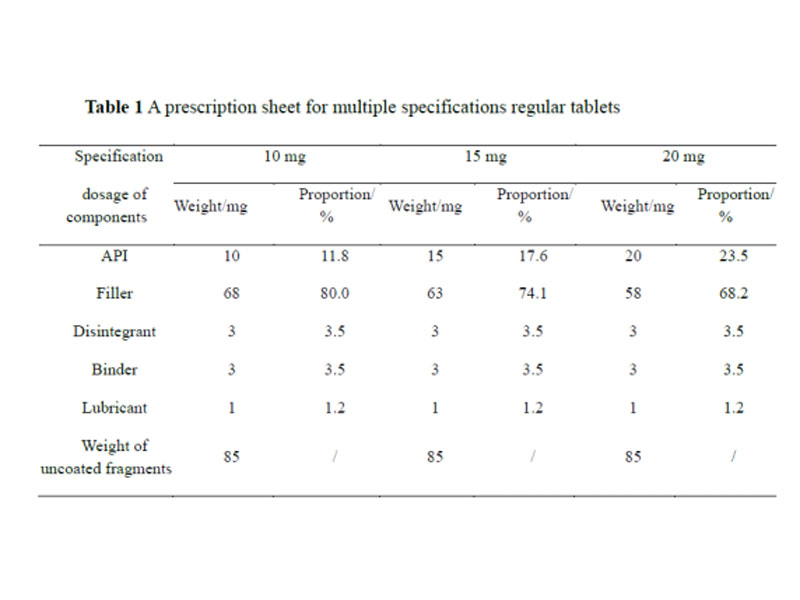
The 20 mg strength is the BE strength, and a BE exemption for the 10 and 15 mg strengths is proposed. The active ingredients of the three specifications are all greater than 5%, and they are not “highly active drugs”, so they are judged according to the standard of “the prescription ratios of the active ingredients and inactive ingredients changing in non-equivalent proportions are similar”. The only inactive ingredient with different proportions in the three formulations is filler. The variation of inactive ingredients between 10 mg and BE is 11.8%, which is beyond the range of ±10%, and does not belong to the similar proportions of the formulation, while the 15 mg and BE have similar proportions. The variation in the inactive ingredients of the specifications was 5.9%, which is a similar proportion of the prescriptions.
In Vitro Studies Of Reference Product Varieties Cannot Be Recommended
There are many situations in which reference preparations cannot be recommended, such as varieties with unclear clinical value, unique domestic varieties with unclear information on original research products, and original research products that are not suitable as reference preparations due to discontinuation of production or due to quality and other reasons, and there is no internationally recognized equivalent. Varieties of drugs, varieties that have been assessed by the State Drug Administration as having problems in safety, efficacy and quality controllability, etc. In cases where reference preparations cannot be recommended, some varieties have clear clinical value. The National Center for Drug Evaluation has compiled and released the “List of Chemical Drugs with Clear Clinical Values and Unable to Recommend Reference Preparations (First Batch)” (Draft for Comment) [24], which contains nearly 30 kinds of oral solid preparations, such as calcium gluconate tablets, aluminum and magnesium chewable tablets, and acetaminophen soft capsules. On this basis, the “Technical Requirements for Pharmaceutical Research of the First Batch of Chemical Drugs Not Recommended for Reference Preparations” (Draft for Comment)[25] was drafted, including 13 oral solid preparations. For such drugs, in addition to not requiring consistent evaluation of the quality and efficacy of reference preparations, quality improvement studies are required to refer to the current national pharmacopoeia standards, technical requirements for pharmaceutical research and relevant guidelines. For varieties that have not been marketed in China, research should be carried out in accordance with the technical requirements of innovative drugs, and clinical effectiveness trials should also be conducted under special circumstances.

Summary
In vitro consistency evaluation of oral solid generic drugs is an important part of quality consistency research. In addition to routine investigation items and dissolution curve similarity research, some unconventional items need to be studied based on product characteristics, such as functional scoring, ethanol induced dose dumping, nasogastric tube administration and phosphorus binding capacity. Combined with specific cases, this paper summarizes and analyzes the common problems and concerns in unconventional projects, the Similarity Study of dissolution curve and the quality improvement of generic drugs that cannot recommend reference preparations, and puts forward corresponding solutions and treatment suggestions based on the current regulatory guidelines and technical requirements. With the progress of science and technology and the improvement of regulatory requirements, the technical requirements of in vitro consistency evaluation will be constantly updated. Research should be carried out in time to ensure that the whole product life cycle of the product after approval is consistent with the quality of the reference preparation.
