






1 Overview Of Cleaning Verification And Four Stages
In pharmaceutical enterprises, the same equipment may be used for the production of multiple products, and after the end of drug production, effective cleaning of the relevant equipment used in production is a necessary means to prevent drug contamination and cross-contamination. The provisions on cleaning and preventing cross-contamination have always been emphasized in the GMP clause, and as early as 1963, the United States promulgated the GMP Regulation (133.4 It is written that “production equipment must be kept clean and orderly.” In order to meet the requirements of relevant laws and regulations, pharmaceutical manufacturers should ensure that the residue of the product can be removed from the surface of the equipment through certain cleaning procedures, and provide written evidence to prove that various pollution and cross-contamination have been effectively prevented. The cleaning procedure of the equipment depends on the nature of the residue, the structure of the equipment, the material and the method of cleaning, for the determined equipment and products, the cleaning effect depends on the cleaning method, and the written and determined cleaning method is the so-called cleaning procedure. The operating parameters of the cleaning process include various parameters such as the type of cleaning agent, concentration, contact time, temperature and so on. In the pharmaceutical industry, the concept of cleanliness refers to a state in which the total amount of various residues in the equipment, including microorganisms and their metabolites, is as low as not affecting the prescribed efficacy, quality and safety of the next batch of products. Through effective cleaning, the substances left in the production equipment of the previous batch of production can be reduced to the extent that they do not affect the efficacy, quality and safety of the next batch of products. Cleaning verification is to confirm the effectiveness of cleaning procedures, and collect sufficient evidence through scientific methods to confirm that the equipment after cleaning according to the prescribed method can consistently meet the predetermined cleaning standards. The common practice is to divide the cleaning verification into four stages, the method development stage, the program preparation stage, the program implementation stage, and the verification status maintenance stage, And Figure 1 processes each stage, which is elaborated below.

1). The Development Phase
According to the nature of the product, the characteristics of the equipment, the production process and the raw materials and accessories used and other factors for laboratory simulation, the development of cleaning methods and the development of cleaning procedures, cleaning personnel for operation training.
2). Scenario Preparation Phase
First of all, a cleaning verification plan should be prepared, the design and strategy of cleaning verification should be listed, the production equipment should be examined in detail, and representative and difficult-to-clean parts should be identified as sampling points; Calculate the surface area of the equipment, select a certain substance as a reference substance according to the relevant nature of the product, determine the maximum residue allowed after cleaning as the qualification standard, determine the degree of cleanliness of the equipment by testing its content during verification, and if necessary, check the residual amount of the detergent; Development and approval of verification schemes in accordance with common requirements for verification, development of sampling and testing methods related to verification to ensure the accuracy of the data, and training of relevant personnel before verification begins.
3). Programme Implementation Phase
Trials were conducted to obtain data in accordance with the approved validation protocol, and the results of the evaluation were concluded. If the verified results indicate that the cleaning procedure does not ensure that the equipment is cleaned to the predetermined standard, it is necessary to find the cause, modify the procedure and re-verify until the result is qualified.
4). Validation Status Maintenance Phase
The cleaning method that has been verified is immediately carried out in the maintenance stage, the cleaning method that has been put into operation is monitored, the change management of the cleaning method is implemented, and the actual effect of the cleaning method used in various production activities can be seen according to the results of the monitoring, so as to determine the revalidation cycle for revalidation.
2 Design And Development Of Cleaning Process
The cleaning process can be divided into physical methods and chemical methods, physical methods include showering, scrubbing, vacuum dust removal, the use of physical cleaning methods must consider the solubility of residues, batch size, and the degree of adhesion on the surface of the equipment. Chemical cleaning mechanisms include dissolution, emulsification, wetting, chelating, dispersion, hydrolysis, oxidation, etc.
The common processes of residue contact with the cleaning liquid, wetting, and detachment from the surface of the equipment are discussed in detail here using the most common cleaning mechanism, dissolution, as an example.
The cleaning process with the mechanism of dissolution is mainly through the dissolution of the solvent on the residue and the impact of the flowing cleaning liquid on the residue so that the residue attached to the surface of the equipment enters the solvent. Microscopically, the speed of dissolution depends on the difference between the number of solute molecules entering the solution from the solute surface and the number of molecules returning to the solute surface from the solution in a unit time. Once the difference is zero and the surface dissolution process reaches dynamic and smooth, this solution is a saturated solution. During the dissolution process, a thin layer of saturated solution is quickly formed from the surface of the solute, and the solute molecules in the saturated solution continue to diffuse deep into the solution, forming a decreasing concentration gradient from the surface of the solute to the depth of the solution. If the solute molecules in the saturated layer cannot quickly enter the depths of the unsaturated solution, the rate of dissolution is reduced. Therefore, even substances with a large solubility, such as the bulk crystals of sucrose (commonly known as rock sugar), are very slow to dissolve in the static state without stirring, and the way to improve the dissolution rate is to increase the flow rate of the solution.
During the cleaning process, the cleaning agent must be brought into contact with the residue during the movement. The relative motion of the cleaning agent and the residue can be decomposed macroscopically into vertical and horizontal movements. Relative motion can quickly take dissolved substances away from the surface of the solute, while relative motion in the horizontal direction can be divided into two types of cases, laminar flow and turbulence, according to the basic principles of fluid mechanics.
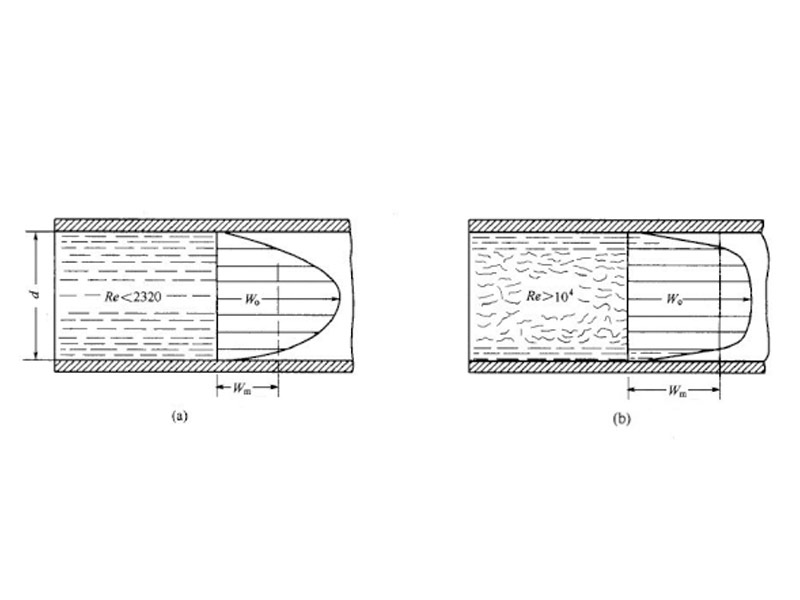
Figure 1 Flow Rate Of Fluid In Laminar And Turbulent Flow
(a) Laminar flow state; (b) Turbulence status
W0—maximum flow rate; Wm—Average flow rate
Laminar flow is when a fluid flows through a catheter, where all particles flow in a square parallel to the tube shaft. At this time, the velocity of the fluid is the largest at the axis of the pipe, and the velocity from the axis to the wall of the pipe is gradually reduced to equal to zero. It can be inferred from this that if the cleaning agent forms a laminar flow in the equipment to be cleaned, it will quickly form a stable saturated solution layer on the surface of the residue, and the dissolution rate of the residue will drop sharply, which is very similar to the dissolution process in the stationary state, so that the cleaning efficiency will also be significantly reduced. Therefore, laminar flow should be avoided in cleaning. When the fluid flows in the form of turbulence, although the fluid flows in one direction along the pipe on the macroscopic level, the speed of motion of each particle changes at any time in size and direction from the microscopic point of view. There are always some particles that move in a direction relatively perpendicular to the tube shaft or tube wall. In this way, a stable saturated layer will not be formed on the surface of the residue, the speed of dissolution will be greatly increased, and the efficiency of cleaning will also be improved. Therefore, during the cleaning process, it must be ensured that the cleaning liquid flows in the form of turbulence, and the form in which the fluid flows depends on the size of the re of the reault coefficient of the fluid.
In the Re=dωρ/μ formula, d is the pipe diameter; ω is the flow rate; ρ is the fluid density; μ is viscosity.
当Re<2300when laminar;Re>10000when turbulence;2300<Re<10000, is the transition phase of laminar and turbulent flow.ReThe larger the surface turbulence, the more intense the turbulence, that is, the greater the change in the direction and rate of movement of the particles, and the faster the residue dissolves.
In the case of the identified cleaner and eluent, Re is proportional to the product of the pipe diameter and the flow rate;
Re∝dω
The more common clean-in-place process involves the cleaning agent being driven by a pump in the process of circulating between the equipment and the pipe. For the identified system, the clean flow V is fixed. According to the incompressible nature of the liquid, in the absence of parallel pipes and forks, the flow rate at each point in the pipe must be the same, regardless of the change in pipe diameter.
Because V=ωS=ωπR2=π/4×ωd2, where S is the pipe cross-sectional area
Thenω=4/π×V/d2
Then Re∝ V/d, if V is a fixed value,
Then Re∝1/d
It can be seen that in the system, the re value of the part with a large diameter or the part of the pipe diameter from small to large is small, and it is relatively prone to laminar flow and is more difficult to be cleaned. For systems with multiple parallel pipes, especially different pipe diameters, these parts are usually listed as difficult to clean due to the change of flow rate and flow distribution of each pipe. In addition, we must not ignore those parts that do not seem to be in direct contact with the product, such as compound amino acid injection preparation system generally needs to install explosion-proof safety valves (membranes) of manifolds, exhaust pipes, nitrogen-filled tubes, vacuum tubes, etc. These pipes may be contaminated due to the flying of material particles during feeding, or because the atomized droplets in the preparation tank are scattered with processes such as nitrogen filling and vacuuming. Sometimes this contamination is mild, but if the cleaning procedure fails to take these pipes into account, it can have serious consequences over time. In general, all dead corners, parts that are not easy to contact with cleaning agents, such as pipe connections with sealing gaskets, parts with rapid changes in pressure and flow rate, such as manifolds or forks, pipe diameters that change from small to large, and parts that are easy to adsorb residues, such as parts where the inner surface is not smooth, should be regarded as the most difficult to clean parts. The mechanism of emulsification and chemical reaction is similar to the dissolution process at the microscopic level, and there are cleaner molecules acting on the surface of the residue, causing the molecules on its surface to detach or react to form other substances and then dissolve, so the macroscopic parts that are not easy to form turbulence are also difficult to clean.
3 Classification Of Cleaning Methods And Principles Of Cleaning Agent Selection
Classification Of Cleaning Methods
According to the cleaning position, the cleaning method is classified, and the cleaning method can be divided into two categories: online cleaning and offline cleaning. According to the classification of automatic / manual cleaning methods, the cleaning methods can be divided into automatic cleaning and manual cleaning. Manual cleaning To ensure the reproducibility of the cleaning procedure, documentation is required for detailed process descriptions, operator training, adequate monitoring, and a clear written cleaning procedure help ensure consistency in manual cleaning. Automatic cleaning usually does not involve personnel intervention, the cleaning system usually programs different cleaning strokes, and the automatic cleaning method can be used to monitor the stroke and parameters of automatic cleaning consistently and stably.
1). Online Cleaning
Cleaning of large equipment can be carried out at the installation location of the equipment, generally very similar to the layout it is used in production, and in-line cleaning can be an automatic or manual cleaning process. In-line cleaning systems use spray devices to cover the surface of process equipment with cleaning agents and remove residues through physical impact, spray balls can be stationary or moving (e.g., rotating, oscillating), these systems are often used to clean large pieces of equipment such as mixing tanks, fluidized beds, reactors, etc. Solvent reflux cleaning method, some volatile solvents are boiled in the reactor, and when the vapor of the solvent condenses on the surface of the equipment, the residue on the surface can be dissolved. Placebo cleaning method this method needs to choose a placebo that will not adversely affect the quality of the next batch of products, the principle of this method is that when the placebo flows in the equipment, the drug residues and process residues of the previous batch of products will be removed, the advantage of this method is that the placebo in the equipment Processing process is the same as the actual production of the product, so the placebo and the next batch of products in the same contact with the surface, the disadvantage is that the cost is high, and it is difficult to prove the effectiveness of the cleaning process.
2). Offline Cleaning
For equipment parts and portable process equipment that are difficult to clean after installation, they are usually transferred to another designated cleaning room for automatic or manual cleaning after disassembly, manual operation is indispensable in offline cleaning, and generally needs to be described in detail in the document and trained accordingly. Regardless of the cleaning method used, a detailed written procedure must be developed to prescribe the cleaning procedure for each piece of equipment, so as to ensure that each operator can perform the cleaning in the same way and obtain the same cleaning effect. This is a prerequisite for cleaning verification. In order to ensure the reproducibility of the cleaning and the reliability of the verification results, the cleaning procedures should at least stipulate the following:
- Necessary disassembly requirements for the equipment before the start of cleaning and assembly requirements after cleaning;
- The name and main ingredients of the detergent used;
- Preparation method of detergent;
- Key parameters such as the time, temperature, and flow rate of the cleaner contact surface;
- Leaching requirements;
- The maximum time from the end of production to the startof cleaning;
- The maximum time of continuous production;
- The cleaned equipment is used for the maximum storage time before the next production.
Choice Of Detergents
The cleaning agent should be able to effectively dissolve the residue, do not corrode the equipment, and itself is easy to be removed, with the improvement of environmental protection standards, the cleaning agent should also be required to be as harmless to the environment as possible or can be harmless treatment, to meet the above requirements and should be as cheap as possible. According to these criteria, water is the preferred cleaner for water-soluble residues. From the perspective of verification, the cleaners with different batch sizes should have sufficient quality stability. Therefore, it is not advisable to promote the use of general household cleaners, because of its complex composition, uncontrolled microbial contamination during production, large quality fluctuations, and suppliers do not publish detailed composition. The use of such cleaners also raises another problem, namely how to prove that the residue of the detergent meets the standard. As simple and precise as possible, cleansers should be chosen. According to the nature of the residue and equipment, the company can also formulate its own cleanser with simple and precise effect, such as a certain concentration of acid, alkali solution, etc. Enterprises should have a sufficiently sensitive method to detect the residue of the detergent and have the ability to recycle or treat the waste liquid harmlessly. In general, the cleaning agents used for post-production cleaning are usually divided into four categories.
Water
Water is usually used for the preparation of pre-elution, after elution and dilution of the drug, but for residues that are easy to clean with water, water can also be used directly as a cleaning agent. The water used for cleaning includes tap water, demineralized water, purified water, water for injection, etc. Under normal circumstances, the water quality used for final leaching is at least equivalent to that of drug production water, and the quality of clean water should also meet the chemical, microbial and endotoxin limits for its use.
Organic Solvent
Organic solvents are generally used for cleaning in the synthesis process of APIs, the choice of solvent is based on the solubility of residues in the solvent, unlike water, organic solvents can use solvent reflux cleaning method. However, it has the impact of safety and environmental protection, so the general factory will prefer water as a cleaning agent.
Acids And Bases
The strong acidity and alkalinity of the acid-base solution will promote hydrolysis, hydrolyzing the macromolecular organics, and simplifying the residual structure and easy to remove. Acid-base cleaners have the advantages of a single component, low price and easy removal, but commercially available cleaners, such as sodium hydroxide, have limited cleaning effects on strong adsorption or dry residues, and also have a certain moisture absorption and dirt suspension effect.
Formulated Cleaner
Formulated cleaners contain multiple ingredients, utilize different cleaning mechanisms, and therefore have a wider and more effective cleaning effect, in addition to having the alkaline and hydrolytic effects of commercially available bases, formula detergents may provide better wetting and interactions such as dirt permeability, emulsification, etc.
4 Key Points Of Cleaning Operating Procedures

The degree to which a piece of equipment needs to be disassembled should be specified in the cleaning procedures of the equipment, most equipment, such as a filling machine for large-volume injections, a one-step granulator for solid preparations, etc. needs to be disassembled to a certain extent before cleaning, and the filling machine for small needles can almost be said to be completely disassembled. There should be written, clear and complete dismantling instructions, preferably with schematic diagrams to make them easy for the operator to understand.
Pre-Wash/Inspect
The purpose of pre-washing is to remove large amounts of (visible) residual product or raw material, creating a basically consistent starting condition for subsequent cleaning.
Since cleaning procedures are often not dedicated, it requires common equipment suitable for the production of multiple products and concentration or dosage specifications to simplify management and operation, so pre-washing is required. The role of pre-washing is to establish a relatively consistent starting point to improve the reproducibility of subsequent steps. The water quality used for pre-washing does not have to be demanding, usually drinking water or drinking water purified by a certain procedure (such as filtering) is sufficient, and the equipment is flushed with fresh running water using a water pipe or a handheld high-pressure spray gun to remove residues. For the situation where the physical properties of residues are quite different, some enterprises hope to formulate a comparison table corresponding to the product and pre-washed parameters such as water temperature, pressure, time, etc., and the operator selects the parameters according to the actual product. This approach is not ideal when implemented. Due to the quality and habits of the operator, from a large number of programs to choose the program should be adopted is easy to cause errors, the simpler and practical method is to let the operator check whether there is still visible residue, let them continue to spray the equipment until the visible residue disappears, as the end of the pre-washing. Therefore, the operator’s criteria for judging whether the pre-washing is complete or not must be as clear as possible, especially the parts that should be inspected. For example, it can be stipulated in the regulation that all surfaces of the machine can be continuously sprayed with hot drinking water to make all visible residual particles disappear, with special attention to inspecting parts that are not easy to clean.
Cleaning Parameters
The operating parameters of the cleaning procedure (such as the type of detergent, concentration, contact time, characteristics of residues, contamination conditions), as well as the characteristics of the cleaning equipment, the automated cleaning path, the order of the cleaning environment, and the flow rate of each step need to be confirmed before being put into use. Each step of the cleaning procedure consists of 4 parameters, namely time, action, concentration and temperature. These four parameters are interrelated and have a direct relationship to the success of each stage of the cleaning cycle, such as improving decontamination by heating the cleaner. Variables as cleaning parameters need to be determined, and acceptable ranges of cleaning parameters are established as part of the cleaning program development effort.
Time

Defined as the length of time of the cleaning step, in one cleaning step, it can be defined and measured in two ways: direct and indirect, and the direct method can be used as a timer in the control system to measure time. Time can also be measured by indirect methods, for example during leaching, sometimes by measuring volume instead of measuring time, since time can be determined by volume and flow rate. For the final rinse water, it is common to increase test requirements, such as conductivity.
Action
Fluid action defined as a cleaner. Such as soaking, washing, impact, turbulence. Agitation can improve the effectiveness of the cleaner and the effectiveness of the cleaning process. Typical manual cleaning involves soaking and scrubbing to achieve a cleaning effect. Automatic cleaning procedures usually use shock or turbulence as cleaning actions. The cleaning procedure requires a clear cleaning action. Flow rate is an important parameter of the cleaning agent and cleaning water as it flows through the equipment, and the flow rate should be specified and confirmed at each step of the cleaning process. Spray equipment should have the requirements of maximum and minimum flow, and the elution flow rate of the pipeline should ensure the formation of turbulent flow.
The Concentration Of The Detergent
Directly affecting the success of the cleaning procedure, chemical cleaners can be concentrated after dilution. The cleaning effect is related to the concentration of the detergent, the cleaning agent may not be able to achieve the cleaning effect by using too little, and the residue from the cleaning agent may be difficult to remove after using too much, and a large amount of elution is required. In general, the best cleaning effect for alkaline cleaners can be achieved by increasing the temperature in the agitated state or extending the time of the turbulent leaching cycle. Chemical cleaners generate significant capital investment in both procurement and disposal, so it is extremely important to determine the correct concentration to ensure the cleaning effect. Automatic systems for cleaning agent addition must be reproducible. Regardless of the method of addition, confirming the detergent concentration helps to confirm the consistency of the method. For automatic cleaning procedures, conductivity testing is the easiest way to test the concentration of strong alkali or strong acid cleaners. Abnormal changes in the concentration of the cleaner should be detected online by the chemical composition of the detergent, such as some detergent addition systems are controlled by volume and conductivity testing is used as a confirmation method. Alarms are issued when the conductivity exceeds a preset value, and the allowable range is required from data developed by the cleaning program.
Temperature
The optimal temperature range for different steps in the cleaning procedure will vary, and the typical temperature of the initial cleaner is room temperature, with the aim of maximum removal of denaturation or degradation products and maximum dilution products. The cleaner is heated to improve the effect, and the final cleaning water can be passed at high temperatures to speed up the drying rate and increase the solubility of any process and detergent residues.
5 Clean Verification Master Plans
All validation activities should be planned, the requirements of the clean validation plan should be specified and documented in the master plan or similar documentation, and the content of the master plan for clean validation of pharmaceutical production may in principle be the same. The plan should describe the responsibilities, the clean verification plan, and the implementation of the clean verification. It is best to have a detailed master plan for cleaning validation, which is described in the plant-wide validation master plan, and the master plan for cleaning verification may contain everything. Another approach is to prepare an overview version of the clean validation master plan, followed by a clean validation execution or project plan detailing the requirements for the clean validation. These physically existing documents should be regularly reviewed and updated, and plan reports should be prepared on a regular basis summarizing important activities in the implementation of the plan. The plan should describe every important aspect of the cleaning verification process, the composition of the master plan and the appropriate details that need to be provided depend on the actual operation of the particular facility, and the composition of the master plan includes but is not limited to the following topics.
1). Purpose

The Clean Validation Master Plan helps the company’s management understand the timing of the validation activities, the arrangement of personnel, and the need for related validation and validation activities, helps the validation team members understand their respective responsibilities, and gives the people involved in the project and the inspectors a global view of the verification methods used and all the validation activities used at the plant.
2). Scope
The Clean Validation Master Plan includes provisions on general principles of validation, acceptable standard calculations, grouping (matrix) and worst-case product evaluation.
3). Provision Of Duties
Detailed description of the responsibilities of each department in the verification process.
4). Product And Device Grouping (Matrix) And Worst-Case Product Evaluation
Usually a production line will produce multiple varieties at the same time, each variety consists of active ingredients and excipients, and it is not necessary to set limit standards for all residues and test them one by one in the cleaning verification, because this is impractical and unnecessary. In a sense, the cleaning process is a dissolving process, so the common practice is to determine the most difficult to clean substance from each component, as the target compound is the verification object, the target compound generally takes into account its characteristics, such as:
Solubility risk
Toxicity, pharmacology
It is difficult to clean, such as a certain adhesion to the surface material of the equipment
Products that contain greases, pigments or flavoring agents that are difficult to clean (color, aroma and taste) in the formulation
Varieties with high production volume (products with high production frequency corresponding to high cleaning frequency)
If the cleaning process uses a cleaning agent, its residue should also be regarded as a marker
There may be many types of devices used in the same process, and for the same type of equipment, consider grouping them and validating them at the same time. For device grouping, the grouping principle is defined in terms of form and function, and the devices with similar design and function and different sizes can be divided into a group.
5). Calculation Of Residue Limits And Acceptable Criteria
Acceptable limits for chemical residues
Determination of detergent residues
Determination of the level of microorganisms
How to determine the residue limit is a rather complex problem, and enterprises should formulate scientific and reasonable limit standards that can be achieved and tested by appropriate methods according to the actual situation of their production equipment and products. At present, the generally accepted limit standards of enterprises are based on the following principles: limits based on visual inspection; Acceptable limits for chemical residues; Microbial residues can be limited.
(1) Limits Based On Visual Inspection
Visual inspection requires no visible residue, and inspection is required and the results of the inspection are recorded after each cleaning, which should be the first acceptance standard for the acceptance limits of cleaning verification.
(2) Acceptable Limits For Chemical Residues
There are two ways to calculate acceptable limits for chemical residues, the biological activity limit (1/1000 of the minimum daily dose) and the concentration limit (10 ppm). When considering acceptable residue limits, consider both methods and select the most stringent criteria as the final criterion for cleaning verification.
A) Acceptable criteria for the use of health-based data
When an acceptable daily exposure level (ADE) or permissible daily exposure (PDE) value is available, the maximum allowable residue (MACO) should be calculated based on ADE. The principle of MACO calculation is based on the ADE/PDE value, which calculates the amount of residue that allows to be carried from the previous product into the next product. Calculate the ADE value or PDE value according to the following companies, and use the results for the calculation of the MACO value:
ADE=NOAEL×BW/(UFc×MF×PK)
Calculate the MACO value from the ADE value according to the following formula:
MACO=ADEprevious×MBSnext/TDDnext
ADE —- acceptable daily exposure levels

MACO —- maximum allowable carryover: the maximum acceptable amount of carrying from the previous product to the next product
BW—- average adult body weight, UFc —- component uncertainty factors: reflecting the individual variables, different varieties of differences, subacute conversion to acute extrapolation, the inference of the lowest visible damage level to the level of no visible damage effect, data integrity and other compensation factors
MF—- Correction Factor: Used to express uncertainties that are not covered by other factors
MBSnext —- the minimum batch size for the next product
TDDnext —- standard treatment day dosage for the next product
B) Acceptable limits of biological activity:1/1000of the minimum daily therapeutic dose
Determining the limits of residues based on data on the biological activity of the drug— minimum daily therapeutic dose (MTDD) is a common approach used by pharmaceutical companies. Generally take the minimum daily therapeutic dose of 1/1000 as the residue limit, it can be considered that even if there are large individual differences, the residue will not produce a pharmacological response to the human body. Therefore, drugs with high activity and sensitivity should be used in this method to determine the limit of residues.
The general surface calculation formula is as follows:
L1/1000=MTDa/1000×Nb/MDDb×Sb
MTDa —- the active ingredient content in the minimum daily dose of the product prior to cleaning
Nb —- batch size of the product after cleaning
MDDb —- the active ingredient content of the maximum daily dose of the washed product
Sb—- the percentage of active ingredient content of the product after cleaning (%, W/W).
C) Concentration limit: one in 100,000 (10ppm).
The quantity level of residues in the next product should not exceed one in 100,000 (10ppm), which is based on the analytical method The ability to achieve the ability to develop, from the perspective of controlling microbial pollution and pyrogen pollution, is also relatively safe. In general, unless it is a highly active and highly sensitive drug, the safety of this limit is sufficient. The concentration of residues per unit area (surface residue limits) on the inner surface of the equipment can be derived from the residue concentration limits, assuming that the residues are evenly distributed on the inner surface of the equipment and are all dissolved in the product at the time of the next batch production. The production batch of the next batch of the equipment is B(kg), because the maximum residue concentration is 10× 10-6 that is, 10mg/kg, the total amount of residue is up to 10B(mg);” The limit of the residue per unit area is the total amount of residue divided by the measured inner surface area in contact with the product, and the total internal surface area of the equipment is SA(cm2), then the surface residue limit L=10B/SA (mg/cm2) 。 To ensure safety, the safety factor F should generally be divided by L=10B/(SA×F) (mg/cm2) The elements to be considered in calculating the acceptance residue limit are as follows (Calculating Acceptance Residue Limit Considerations).
| Influencing Factors | Minimum Daily Therapeutic Dose/Toxicity | Solubility | Batch | Maximum Daily Therapeutic Dose | Area In Contact With The Product | Difficult To Clean Locations | Sampling Area |
| Product Before Cleaning | √ | √ | |||||
| Product After Cleaning
|
√ | √ | |||||
| Equipment | √ | √ | |||||
| Sampling Method | √ | ||||||
| Detergent | √ | √ |
(3) Microbiological Limits
The microbiological verification of cleaning can be carried out simultaneously with the chemical verification of cleaning, and the characteristics of microorganisms are rapid reproduction under certain environmental conditions, and the number increases sharply. And the microorganisms present in the air can contaminate clean equipment in various ways. The longer the equipment is stored after cleaning, the greater the chance of contamination by microorganisms. Therefore, enterprises should comprehensively consider the actual situation and needs of their production, and formulate their own limits for microbial pollution level control and the maximum storage period from cleaning to the next production.
(4) Limit Standards For Residual Solvents
Organic solvents other than water may be used in the production and cleaning of pharmaceutical products, and the ICH divides solvents into 3 levels in the Residual Solvent Guidelines.
A) A Class Of Solvents
Because of their unacceptable toxicity or environmental hazards, Class I solvents should not be used in pharmaceutical production, and if they must be used, the residues are controlled as follows, and their names are as follows (Class I Solvent Concentration Limits).
| Solvent | Concentration Limit(ppm) | Remark | Solvent | Concentration Limit(ppm) | Remark |
| Benzene | 2 | Carcinogen | Carbon Tetrachloride | 4 | Toxicity And Harm To The Environment |
| 1,2 – Dichloroethane | 5 | Toxicity | 1,1,1 – trichloroethane | 1500 | Hazardous To The Environment |
| 1,1 – Dichloroethylene | 8 | Toxicity |
B) Class II Solvents
Because its toxicity should be limited in the formulation and the permissible daily intake should be strictly limited.
C) Solvents Of Class III
The third type of solvent is low toxicity, the harm to the human body is very small.
The ICH Guidelines on Residual Solvents stipulate that Class I and Class II solvents are used only in the production of pharmaceutical products in irreplaceable circumstances, but cannot be used as detergents. When unavoidable, class III solvents can be used as cleaning agents, and the residual concentration of the solvent allowed in the next batch production should not exceed 0.5% of the initial solvent concentration.
6). The Most Difficult Parts To Clean And The Sampling Point
The most difficult parts of the equipment to clean are generally determined according to the production experience and risk assessment, taking into account the area that cannot be covered by on-line cleaning and the surface of the parts that cannot be disassembled in manual cleaning, in addition, for different components of the equipment, the different materials used, it is necessary to consider them comprehensively to confirm the most difficult parts of the equipment to clean. Identifying the most difficult parts to clean can first be used as a reference in equipment design, and secondly, to further confirm the sampling point in the cleaning verification, but this process is only suitable for the method of elution sampling.
7). Clean Verification Sampling Method Confirmation
For confirmation of the sampling method of cleaning difficult clearance, please refer to the sampling method verification below.
8). Other Elements Of the Clean Verification Master Plan Include, But Are Not Limited To, The Following
Clean Verification Time Schedule Cleaning Verification Routine Monitoring/Maintenance Requirements
6 Clean Verification Risk Assessment
Risk Assessment
Risk assessment runs through the entire life cycle of the product, the same applies to the cleaning verification process, the rational use of risk assessment in the cleaning verification can achieve the following purposes:
(1) Select the target products for cleaning verification, effectively reduce the workload of cleaning verification, and improve the applicability of cleaning verification;
(2) Select the appropriate cleaning agent to achieve the purpose of cleaning more efficiently and cost-saving;
(3) Select the appropriate testing method, such as exclusive and non-exclusive methods for different processes and equipment;
(4) Select the location of the sampling point and select a reasonable sampling method to carry out cleaning verification more efficiently.
Cleaning and cleaning verification can be carried out from the relevant process system knowledge, contaminants and equipment cleaning auxiliary systems for risk assessment, confirmation. These systems are subject to design review and then confirmed according to the relevant acceptable standards to demonstrate that the relevant requirements of the system have been met. In the process of cleaning verification, there are many factors affecting the success of cleaning verification, each factor has different potential risks, each factor must be fully analyzed and evaluated to ensure that the cleaning verification is carried out smoothly, the following figure uses fishbone diagram analysis to confirm all influencing factors.
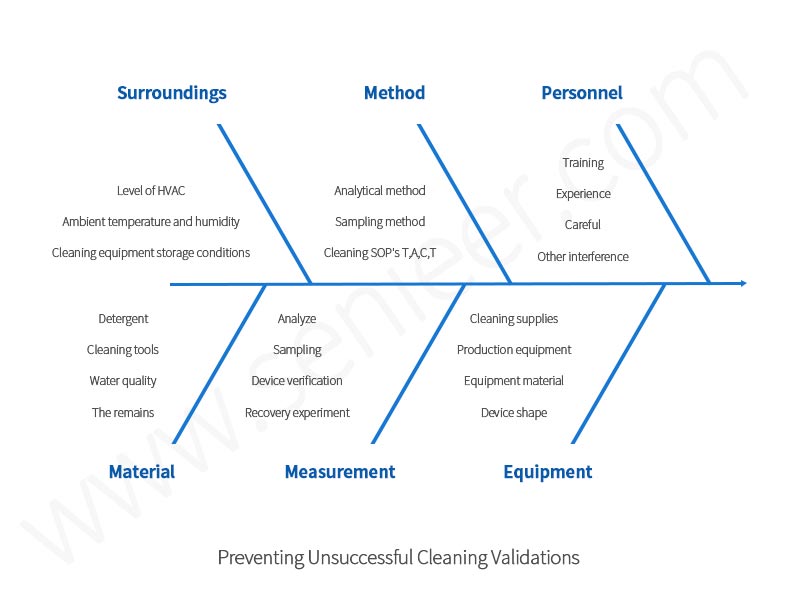
Figure 2 Risk assessment of fishbone diagram
1). Environment
The influence of environmental factors on cleaning verification is crucial, and a good environment can ensure that cleaning verification is carried out smoothly. Environmental factors seriously affect the microbial residue project in the cleaning verification process, different levels of environment have different microbial and dust particle requirements, before the cleaning verification, it is necessary to ensure that the performance confirmation of HVAC has been completed, and the temperature and humidity of the environment have met the requirements. In particular, for the storage conditions of the cleaned equipment, the cleaned equipment must be stored in a dry environment, and the verification of the clean equipment retention time and the dirty equipment retention time is required, and these two time verifications are mainly for the microbial residue limit, because environmental factors if they cannot be effectively controlled, it will inevitably lead to the failure of the cleaning verification.
2). Methods
Before the cleaning verification is performed, the verification of the analytical methods and sampling methods related to the cleaning verification and the cleaning SOP of all relevant equipment must be completed. T.A.C.T (Temperature, Action, Concentration, Time) parameters must be clearly described in the equipment cleaning SOP to ensure the operability of the cleaning procedures.
3). Personnel
For the relevant personnel involved in the cleaning verification, especially the operators of the equipment cleaning related to the cleaning verification, it is necessary to carry out strict training on the relevant cleaning procedures to ensure the consistency of the equipment cleaning, and if necessary, different team members can be used to clean the equipment during the cleaning verification process, so as to prove the durability of the cleaning SOP. The personnel who perform the cleaning verification must all pass the training of the cleaning verification program, and in the implementation process, try to select experienced personnel, especially the sampling operators, and must pass the training of the recovery rate experiment, otherwise the sampling operation cannot be carried out.
4). Material
In order to make the cleaning of the equipment to a certain degree of cleanliness, the cleaning of the equipment must strictly use cleaners and cleaning tools, the cleaning agent must use a single ingredient and the cleaning agent allowed by the pharmaceutical industry, and in the process of cleaning verification execution, the cleaning agent must be determined, the cleaning tool must choose a cleaning tool without any shedding substances, and the change of important cleaning tools may lead to the re-verification of the cleaning procedure. The quality of the water used for equipment cleaning has a great influence on the development of finally acceptable standards, and different water quality cleaning represents different cleaning requirements, such as water for injection is generally a sterile pharmaceutical plant The cleaning of the equipment by purified water is generally carried out in non-sterile pharmaceutical plants, so the quality of the final cleaning water determines the principle of cleaning verification microbial limits. In the process of drug production, each company’s workshop will have many varieties and dosage forms of drugs, due to the cleaning verification process, to consume a lot of manpower and material resources, we can not be for each variety to be separate cleaning verification, in order to reduce costs and simplify the complex cleaning verification, we need to classify all varieties and dosage forms in the workshop, from which to select the worst conditions of the product for cleaning verification.
5). Measure
All equipment instrumentation involved in the clean verification process must be calibrated to ensure the accuracy of the obtained data. Considering the differences in the operation of different personnel, the sampling operation should be carried out by personnel who have undergone strict training and can strictly abide by the procedures, and in order to ensure that the sample has good reproducibility, the sampling operation should be operated by the personnel who complete the recovery rate experiment. The swab is pre-washed with a sampling solvent before use to prevent fibers from remaining on the surface of the sampling location. The recovery rate experiment of different materials must be completed before this protocol is carried out, and should be performed by the same person at least 3 times, which should be greater than or equal to 50%, and the RSD of the three results should not be greater than 20%. In order to ensure the safety of the product, the minimum recovery value should be substituted when calculating the residual amount, that is, the maximum possible residual amount is calculated. Compare the recovery results of different materials, and in order to minimize the risk of contamination, the material with the lowest recovery rate is used as the final recovery rate.
6). Devices
Pharmaceutical production of each company has a different dosage form, each dosage form of the use of equipment is also different, and there are different products, in the implementation of the cleaning verification process, it is impossible to verify the equipment chain of each product, if a company has a certain dosage form of the product is very much, then the cleaning verification cycle will be very long, waste a lot of manpower and material resources. So we will be based on the product use of the equipment chain and the similarity of the product group verification of the device chain, for the same category of equipment chain, only need to select the worst condition device chain verification, as long as the worst condition device chain through the verification, then the rest of the equipment chain does not need to be verified, greatly reducing the burden of clean verification. In the pharmaceutical production process, because there are many types of equipment, each equipment has a different geometry, so the selection of equipment sampling point is very important, the selected sampling point must be strongly representative, and the final sampling point result of the qualification proves that the cleaning procedure of the equipment is applicable.
Because the main purpose of the cleaning verification process is to prove that the active ingredients of the previous batch of products have not caused pollution to the next batch of products, so for some equipment that does not contact the active ingredients, we can appropriately formulate their test items, and not all test items are set in stone
7 Clean Verification Analysis Method Verification
Cleaning verification related analytical methods generally include analytical method verification and sampling method verification, cleaning verification to choose a suitable analytical method is very necessary, the appropriate analytical method should be able to adequately detect the relevant residues. It is also important to draw what conclusions can be drawn from the results of the analysis (e.g. whether the product or detergent was detected, whether the results are qualified), and the results determine whether the cleaning method is feasible or needs to be further improved. This chapter discusses how to select an appropriate test method, including information on suitability and chemical and microbiological test methods, as well as verification of test methods.
Validation Of Analytical Methods
1). Exclusivity
Exclusivity refers to the ability of the analyte to be accurately and reliably detected in the presence of interference from other substances. For cleaning verification, interferences include degradation, excipients, sampling solvents, sampling cotton swabs, cleaners and other interferences.
2). Accuracy
Accuracy is also known as authenticity, refers to the degree of similarity between the true value or the approved reference value and the measured value, generally expressed in terms of recovery rate, the comprehensive recovery rate is required to be no less than 75%, the RSD requirement for the recovery rate is not more than 10%, and the lowest value of the measured recovery rate is used as the correction factor f , the measurement result of the sample divided by the correction factor indicates the actual amount of the sample.
3). Precision
Refers to the degree of proximity of a series of test results for multiple sampling of homogeneous samples under specified conditions, and the precision can be examined from three levels: repeatability, intermediate precision, reproducibility, and the precision investigation should use a homogeneous, credible sample, if not obtained, the sample solution can be artificially prepared for study.
4). Linear
Linearity of the analytical method refers to the ability to proportional the detection result to the concentration (amount) of the analyte in the sample within a given range. Range refers to a certain degree of accuracy, precision and linearity, the test method is applicable to the high and low limit concentration or amount of the analyte in the specimen.
5). Scope
The range of the analytical method refers to a range of higher and lower concentrations of the analyte in the sample, and it has been confirmed that within this interval, the method has suitable accuracy, precision and linearity. Concentrations should range from 50% to 150% of the residue limit.
6). Detection Limits And Quantitative Limits
The detection limit refers to the minimum amount of analyte in the sample that can be detected, and the quantitative limit refers to the minimum amount that can be quantitatively detected in the sample. Detection and quantification limits can be verified using intuitive evaluation and signal-to-noise ratio methods, or using the standard deviation and slope of the response value.
7). Durability
Durability refers to the ability to measure unaffected when the test parameters are appropriately changed slightly, and can be used to illustrate reliability during normal use. Durability test is to change the applicability of the analysis method under the condition of changing its analytical parameters, taking the HPLC method as an example, the typical parameters of the change are as follows: mobile phase PH value change, mobile phase component change, column temperature and flow rate change
8). Solution Stability
Samples placed under certain storage conditions prior to analysis may degrade and change, in which case the stability of the analyte needs to be examined. Using the same solution, the difference in results over pre-selected time intervals is determined to analyze the stability of the sample.
9). System Suitability
During quantitative detection, 2 copies of the control should be prepared, the consistency check of the control is not more than 5%, and the RSD of the control is not more than 10%.
Table 1 Limit checks and quantitative checks of the parameters to be verified in the test method
| Check the method parameters | Limit check method | Quantitative examination method |
| Exclusivity | √ | √ |
| precision | × | √ |
| Linear/range | × | √ |
| accuracy | √ | √ |
| Detection and quantification limits | Detection limit validation | Quantitative limit verification |
| Stability of the sample solution | √ | √ |
| System suitability | √ (one standard) | √ (two standards are parallel) |
Sample Recovery Study
Sample recovery studies often require proof that appropriate analytical methods and sampling procedures are used to adequately measure or quantify residues on the surface of the equipment. These studies provide a scientific basis for the sampling of residue measurements and the establishment of analytical methods. Its purpose is to establish a reproducible device surface recovery rate. Three types of sample recovery are explored below: wiping sample recovery, leaching sample recovery, and “visual inspection” recovery. For wiping and leaching sampling, recovery studies can be used as part of the verification of analytical methods, or can be conducted separately once it is determined that the analytical method can detect residues in the solution. The sample recovery study is a laboratory study that requires specimens of different equipment materials (e.g. stainless steel, glass, PTFE and EPDM) to be sampled and coated with residues to be tested.
1). Wiping Recovery Rate
In the study of wiping recovery rate, the known concentration of residue solution is evenly coated on the material specimen, naturally dried, sampling is carried out by a certain wiping method, the appropriate solvent is selected to extract the residue on the cotton swab, and then the number of residues in the extract is detected. The ratio of the amount recycled to the amount added to the material specimen is the percentage recovery rate of the sample. Since wiping is a manual operation, it is usually necessary to repeat the recovery rate study three times per person. Each combination of residue and surface type requires at least two people to conduct a wiping recovery study. The recovery rate established by the study can be defined in different ways, but is usually defined as the minimum average recovery rate for any one wiper. An acceptable swab recovery rate depends on how the swab recovery test is performed. If the residue limits or analysis results are not corrected when the recovery study confirms the sampling method, the recovery rate is usually required to be 70% or higher. If the recovery rate is used to correct residue limits or analysis results, the recovery rate is generally 50% or more.
2). Elution Recovery Rate
The study of the recovery rate of the elution method is similar to the recovery rate of wiping, and the target residue solution is coated on the material specimen to dry naturally. For the recovery rate of wiping samples, the predetermined wiping steps must be strictly enforced. In contrast, for leaching sampling, the leaching step cannot be repeated accurately in the laboratory (except in special cases of extraction). However, it is feasible to simulate a leaching procedure in the laboratory. Where possible, the conditions under which the simulated elution should be simulated should be the same as those of the actual equipment, including the leaching solvent and the choice of the leaching solvent temperature. In other cases, the same or worst leaching conditions should be selected as the equipment leaching, e.g. the ratio of the amount of solvent to the surface area to be sampled in the recovery study should be the same or lower as when the equipment was leached.
3). Visual Inspection Recovery Rate
This process is actually the determination of a quantitative “visual inspection limit”, and if the visual inspection is only as a supplement to wiping or flushing sampling, the “visual inspection limit” can be determined without requirement. Visual inspection limits under specified observation conditions can be determined by coating the equipment material specimen with residues of different concentrations (μg/cm2). A trained group of observers is required to determine the minimum residue level when surface residue is clearly visible. The significance of the visual inspection limit is that if the surface of the equipment is determined to be visually clean under the same (or more stringent) observation conditions in the cleaning verification scheme, the actual residual level can be considered to be lower than the visual inspection limit. Appropriate viewing conditions include distance, lighting, and viewing angles. Visual inspection limits depend on the nature of the residue, the surface properties (e.g., stainless steel to PTFE), and the viewer’s vision. The typical visual detection limit reported in the literature is 1 to 4μg/cm2. For visual inspection limits, there is no need to determine the recovery rate. The purpose of the study was to determine a residue level when the residue was clearly visible, so that the residue level on any visually clean surface was below the visual detection limit.
4). Bioload And Endotoxin Sampling Recovery Rates
For microbial sampling, recovery studies are not appropriate to determine surface recovery, one of the reasons is that the counting problems of microbial detection are usually counted in “colony-forming units” rather than individual microorganisms. The second reason is that in a standard sample recovery study, microorganisms die or lose their viability when the material specimen is dried. The third reason is that it is not clear which microorganisms to choose for recovery studies. The fourth reason is that biological loading limits are usually significantly lower than levels that can affect product quality or process performance (e.g. in-line sterilization), so even if the recovery rate is low (< 50%), the introduction of recovery factors does not affect product quality and/or process performance.
8 Clean Verification Program And Verification Status Maintenance
The Cleaning Verification Scheme Refers
To the substance or the most difficult to clean substance, the most difficult to clean parts and the sampling site, how to develop the verification of the qualification standard, that is, the maximum allowable residue cleaning verification program must meet the common requirements of the general verification program. The most critical technical issues in the validation scheme are how to determine the limits and what means to accurately quantify the amount of residue, which includes the development and validation of sampling methods and detection methods.
1). Reference Substances Versus The Most Difficult To Clean
General pharmaceuticals are composed of active ingredients and excipients. For compound preparations, multiple active ingredients are contained. Residues of all these substances must be removed. Is it necessary to set limit standards for all residues to be tested one by one in the cleaning verification? This is impractical and unnecessary. In a certain sense, the cleaning process is a dissolving process, so the usual practice is to identify the most difficult to clean (dissolve) substances from each component as a reference substance. Often, people pay more attention to the residue of the active ingredient than the excipient, because it may directly affect the quality, efficacy and safety of the next batch of products. Therefore, the residue limit of the active ingredient must be one of the criteria for verification of conformity. For example, when there are more than two active ingredients, the most difficult to dissolve of which can be treated as the most difficult to clean substance. Take the compound 18-amino acid injection as an example, it has 18 kinds of amino acids, all of which are active ingredients. The most difficult to dissolve is cystine, which is only slightly soluble in hot water, so it can be used as the most difficult to clean. In this way, the cleaning verification finds residual “references” regardless of other soluble components.
2). The Most Difficult Parts To Clean And The Sampling Point
Sampling points should include the most difficult parts to clean, as well as a variety of materials to consider.
3). Clean Verification Protocol
Clean verification schemes are available in a variety of formats, and common requirements include the following:
(1) Purpose: To clarify the equipment and cleaning methods to be verified.
(2) Cleaning procedures: The SOP of the cleaning method to be verified, that is, the cleaning procedures, should be determined before the verification begins, and the cleaning procedures are listed in the verification plan to indicate that the cleaning procedures have been formulated.
(3) Verifiers: Make a list of participants, explain the departments to which the participants belong and their respective responsibilities, and the training requirements for relevant operators.
(4) Determination of references and limit standards: In this part, the basis for determining reference objects should be elaborated in detail, and the calculation process and results of determining limit standards should be determined. Generally, the list of relevant equipment can be calculated, the total surface area, and the special surface area can be calculated; The list of relevant products, including the main active ingredients and their related physico-chemical properties, MTDD values, etc., determine the reference substance, calculate the limit standard.
(5) Inspection methodology: This part should explain the sampling methods, tools, solvents, main inspection instruments, sampling methods and verification of inspection methods.
(6) Sampling requirements: use schematic diagrams, text, etc. to indicate the specific location and sampling plan of the sampling point, and clearly stipulate when, where, how many samples to take, and how to mark each sample. The content of this section is essential for the implementation of the programme and for ensuring the objectivity of the verification results.
(7) Reliability judgment criteria: In this part, it should be specified that the number of times the verification test must be repeated to prove the reliability of the cleaning procedures to be verified, generally at least 3 consecutive tests, and all data meet the limit standards.
Maintenance Of Clean Verification Status
1). Daily Monitoring
After the completion of the cleaning verification, the cleaning method takes effect, the cleaning method enters the maintenance and re-verification stage, and the enterprise should continuously maintain the cleaning verification state to ensure that the cleaning method is continuous and reliable.
The purpose of monitoring cleaning methods in daily production processes is to further examine the reliability of cleaning procedures, and monitoring is particularly important for manual cleaning processes, as their reproducibility depends to a large extent on the training of personnel and the practical skills of operators.
The monitoring method is to conduct regular sampling tests during daily cleaning to confirm the effectiveness of the cleaning process and the reproducibility of the operation. Review of daily monitoring data to determine the cycle of revalidation.
2). Change Management
When there are any changes to the equipment and cleaning procedures that have been verified, as well as product prescriptions, the addition of new products, etc., which may lead to changes in cleaning procedures or equipment, the relevant personnel should be organized to evaluate the changes to determine whether revalidation is required, and when one of the following circumstances occurs, the cleaning procedure must be revalidated.
(1) When the cleaning procedure changes
(2) When the cleaning agent changes
(3) When there is a major change to the equipment
(4) When adding products that are relatively more difficult to clean
3). Optimization Of Cleaning Methods
In actual production, it is very common for one (group) of equipment to be used for the production of multiple products. Sometimes the physical and chemical properties of various products vary greatly. This raises the question for cleaning protocol makers: Should a separate cleaning protocol be established for each product? Experience has taught us that it is not advisable to develop multiple cleaning procedures for a single piece of equipment: not only because the workload of performing difficult cleaning verification for each procedure is too large, but more importantly, it is easy for the operator to choose the appropriate cleaning method among multiple procedures. A more feasible approach is to select the most difficult to clean product as the control product of all the products involved, take the minimum allowable residue of all products/raw materials as the criterion (worst condition), and optimize the design of cleaning procedures sufficient to remove the product/raw material to reach the residue limit. Validation is directed at the program, which can be applied to the cleaning of all products as long as it is proved to meet the predetermined requirements. Of course, from the perspective of environmental protection and cost savings, if the practice proves that the cleaning procedure is too wasteful for most products, another product can be selected for the above-mentioned procedures to develop and verify. This is the product to which products the method applies to must be very clearly defined in the protocol, and it must also be clear what necessary measures need to be taken to prevent errors in the selection.
The selection principle of the reference product is as follows:
(1) List all products.
(2) Determine a number of physical and chemical properties of the product as evaluation items: such as the solubility of the main active ingredients, viscosity, adsorption, etc., of which the most important property is solubility.
(3) Each product is scored for its evaluation. If the solubility is divided into 1/2/3/4 levels, it means insoluble/slightly soluble/soluble/easily soluble.
(4) According to the experience and nature of the product, formulate appropriate types of detergents.
(5) Calculate the maximum allowable residue limit for each product.
(6) Sort the table from smallest to largest solubility, and select the product with the least solubility as the reference product.
(7) If the cleaner used in the table can be divided into two categories: water/water soluble cleaner (including acid and alkali solutions) and organic solvents, a reference representative product should be selected separately.
(8) The value with the lowest permissible residue limit in the table is determined as the permissible residue limit criterion for the verification scheme.
- Identify the cleaner corresponding to the reference product as the cleaning agent used in the cleaning method.
A statistical table of equipmentproduction products (sorted by solubility)
| Product Name | Active Ingredient Solubility | Suitable Cleaning Agent | Allowable Residue Limit μg/cm² |
| Product A | 1 | 1%NaOH Hot Water Solution | 1.5 |
| Product B | 1 | 95% Ethanol | 2.5 |
| Product C | 1 | 70% Ethanol | 3 |
| Product D | 2 | Hot Water | 2.5 |
| Product E | 3 | Water | 2 |
| Product F | 4 | Water | 1 |
Can be seen from the table, products C, D, B, E, can be cleaned with water-soluble cleaners, the most insoluble product C should be selected as the reference product, cleaning agent 1% NaOH hot water solution, the allowable residue limit is set at 1.5μ g/cm2。 Product A, hex with ethanol as a cleaning agent, can be counted as another category, should choose product A as the representative product, the cleaning agent is 95% ethanol, the allowable residue limit is set at 2.5μg /cm2. The cleaning method is designed according to the condition of the equipment, the established cleaning agent and the residue limits. Cleaned and verified in accordance with the law after production.
The cleaning verification test is carried out at least 3 times, and after each batch of production, it is cleaned according to the cleaning procedures, and the cleaning effect is checked according to the verification plan, and the sampling is taken and inspected. Repeat the above process 3 times, and the results of 3 trials should meet the predetermined criteria. If individual test results exceed the standard, the causes must be investigated in detail. If there is evidence that the results exceeded the standard due to sampling, laboratory errors, etc., this data can be deleted from the statistics. Otherwise, validation failures should be judged. Resampling and re-testing until clean should not be used. Failure to verify means that the cleaning protocol is defective and should be modified according to the clues provided by the test results, followed by a new round of verification.
